Jianwei Wei, Liang Zhao, Cheng He, Sijia Zheng, Joost N. H. Reek, Chunying Duan
J. Am. Chem. Soc., (2019), 141(32), 12707-12716
DOI: 10.1021/jacs.9b05351
Abstract
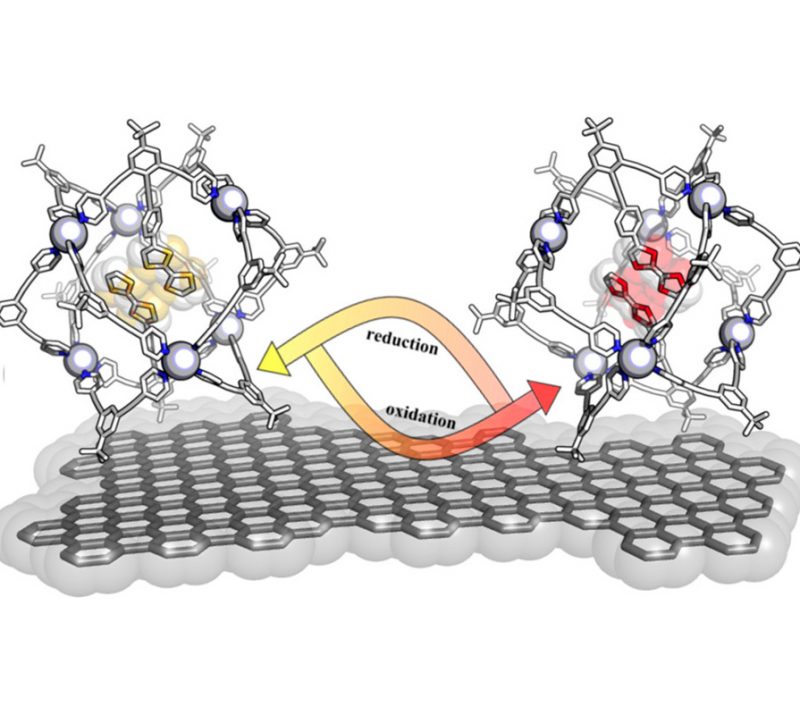
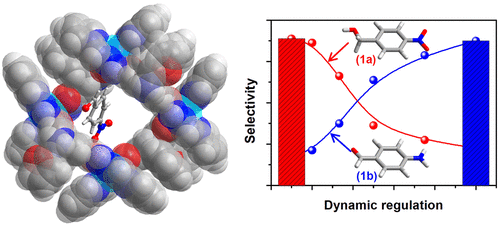
Switchable selective hydrogenation among the groups in multifunctional compounds is challenging because selective hydrogenation is of great interest in the synthesis of fine chemicals and pharmaceuticals as a result of the importance of key intermediates. Herein, we report a new approach to highly selectively (>99%) reducing C═X (X = O, N) over the thermodynamically more favorable nitro groups locating the substrate in a metal–organic capsule containing NADH active sites. Within the capsule, the NADH active sites reduce the double bonds via a typical 2e– hydride transfer hydrogenation, and the formed excited-state NAD+ mimics oxidize the reductant via two consecutive 1e– processes to regenerate the NADH active sites under illumination. Outside the capsule, nitro groups are highly selectively reduced through a typical 1e– hydrogenation. By combining photoinduced 1e– transfer regeneration outside the cage, both 1e– and 2e–hydrogenation can be switched controllably by varying the concentrations of the substrates and the redox potential of electron donors. This promising alternative approach, which could proceed under mild reaction conditions and use easy-to-handle hydrogen donors with enhanced high selectivity toward different groups, is based on the localization and differentiation of the 2e– and 1e–hydrogenation pathways inside and outside the capsules, provides a deep comprehension of photocatalytic microscopic reaction processes, and will allow the design and optimization of catalysts. We demonstrate the advantage of this method over typical hydrogenation that involves specific activation via well-modified catalytic sites and present results on the high, well-controlled, and switchable selectivity for the hydrogenation of a variety of substituted and bifunctional aldehydes, ketones, and imines.
RELATED PROJECTS