Roel F. J. Epping, Felix J. de Zwart, Nicolaas P. van Leest, Jarl Ivar van der Vlugt, Maxime A. Siegler, Simon Mathew, Joost N. H. Reek, and Bas de Bruin
Inorganic Chemistry, 2024, 63(4), 1974-1987
DOI: 10.1021/acs.inorgchem.3c03708
Abstract
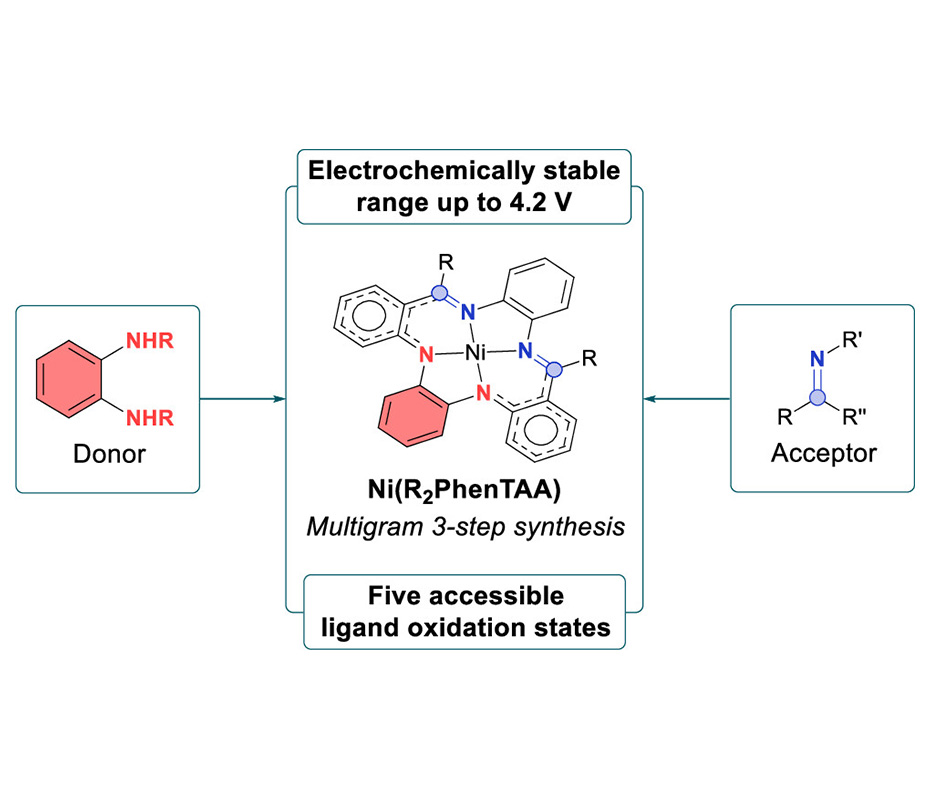
Here, we present the development and characterization of the novel PhenTAA macrocycle as well as a series of [Ni(R2PhenTAA)]n complexes featuring two sites for ligand-centered redox-activity. These differ in the substituent R (R = H, Me, or Ph) and overall charge of the complex n (n = −2, −1, 0, +1, or +2). Electrochemical and spectroscopic techniques (CV, UV/vis–SEC, X-band EPR) reveal that all redox events of the [Ni(R2PhenTAA)] complexes are ligand-based, with accessible ligand charges of −2, −1, 0, +1, and +2. The o-phenylenediamide (OPD) group functions as the electron donor, while the imine moieties act as electron acceptors. The flanking o-aminobenzaldimine groups delocalize spin density in both the oxidized and reduced ligand states. The reduced complexes have different stabilities depending on the substituent R. For R = H, dimerization occurs upon reduction, whereas for R = Me/Ph, the reduced imine groups are stabilized. This also gives electrochemical access to a [Ni(R2PhenTAA)]2– species. DFT and TD–DFT calculations corroborate these findings and further illustrate the unique donor–acceptor properties of the respective OPD and imine moieties. The novel [Ni(R2PhenTAA)] complexes exhibit up to five different ligand-based oxidation states and are electrochemically stable in a range from −2.4 to +1.8 V for the Me/Ph complexes (vs Fc/Fc+).
RELATED PROJECTS